Presented by: Paris Tranos, MD, PhD, ICOphth, FRCS

Edited by: Penelope Burle de Politis, MD

A 21-year-old man presented with subacute visual loss in both eyes.
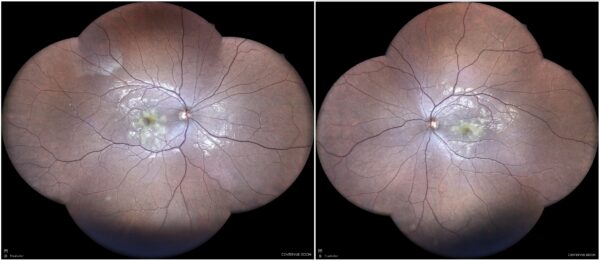
Figure 1: Widefield color fundus photograph (EIDON true-color confocal scanner®) displaying creamy yellowish macular and perimacular lesions in both eyes.
Case History
A 21-year-old, otherwise healthy Caucasian man was referred for investigation of blurred vision in both eyes (BE), more accentuated in the left eye (LE), started 2 weeks earlier. There were no other accompanying symptoms. His past ocular history was unremarkable except for wearing glasses. Upon examination, his best-corrected distance visual acuity (BCVA) was 9/10 in the right eye (RE) and 3/10 in the left eye (LE). Refractometry was respectively +3,00° -1,50°@180° and +3,75° -2,00°@170°. Intraocular pressure was within normal limits bilaterally. Biomicroscopy featured a quiet anterior segment with no cells or flare. Color vision was normal, and there was no relative afferent pupillary defect (RAPD). Fundoscopy revealed yellowish placoid macular and perimacular lesions in both eyes, as evidentiated by widefield retinography (Figure 1). Investigation proceeded with multimodal fundus imaging.
Spectral domain optical coherence tomography (SD-OCT) demonstrated disruption of the retinal pigment epithelium (RPE) and ellipsoid zone at the macula, along with increased choroidal thickness in both eyes, notably on the left. Areas of Henle fiber layer (HFL) hyperreflectivity – known as the angular sign of HFL hyperreflectivity (ASHH sign) – were also noted (Figure 2).
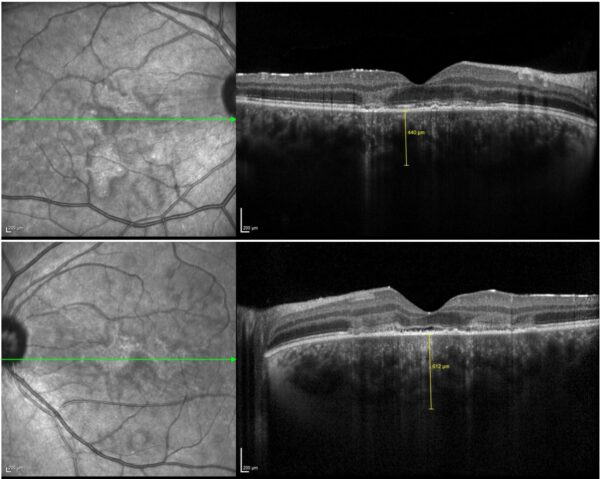
Figure 2: SD-OCT (Heidelberg Engineering®) of both eyes illustrating RPE and ellipsoid zone disruption, ASHH sign, and increased choroidal thickness, especially in the left eye (bottom).
Fundus fluorescein angiography (FFA) showed hypofluorescence of the lesions in the early phase with hyperfluorescence in the late phase (Figure 3).
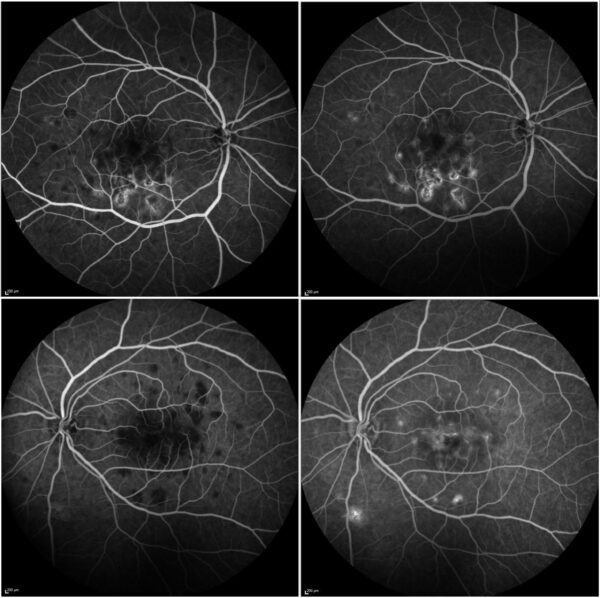
Figure 3: FFA (Heidelberg Engineering®) of both eyes showing numerous posterior pole lesions with early-phase hypofluorescence (left) and late-phase hyperfluorescence (right).
Indocyanine-green angiography (ICG-A) detected multiple areas of hypofluorescence involving the posterior pole and midperiphery of both eyes, more confluent at the macula (Figure 4).
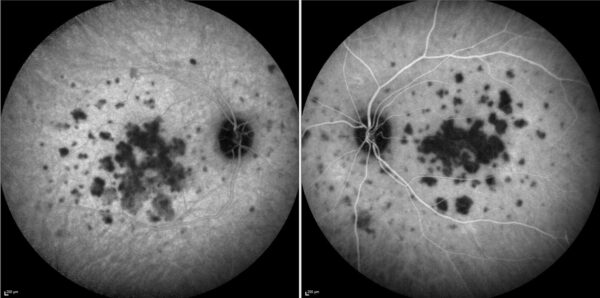
Figure 4: ICG-A (Spectralis, Heidelberg Engineering®) of both eyes showing multifocal hypofluorescent lesions of various sizes, scattered along the posterior pole and midperiphery, confluent at the macula.
Additional History
The patient reported a fever of unknown origin 4 weeks prior, requiring hospitalization and managed with intravenous clarithromycin. At the consultation, he had been afebrile for five days and his blood tests showed no abnormalities.
Based on the clinical presentation and multimodal image findings, the diagnosis of acute posterior multifocal placoid pigment epitheliopathy (APMPPE) was established. A short course of methylprednisolone with tapering was prescribed.
Differential Diagnosis of Acute Posterior Multifocal Placoid Pigment Epitheliopathy
- relentless placoid choroiditis
- macular serpiginous choroidopathy
- multiple evanescent white dot syndrome (MEWDS)
- multifocal choroiditis and panuveitis
- punctate inner choroidopathy
- birdshot chorioretinopathy
- Vogt-Koyanagi-Harada syndrome
- infectious uveitis (syphilis, tuberculosis, fungii)
- vitreoretinal lymphoma
- choroidal metastases
Other white dot syndromes, all characterized by yellow-white placoid lesions with similar imaging and angiographic characteristics, can resemble APMPPE. The differential diagnosis can be made on the basis of uni- or bilaterality, duration, size, aspect, location, distribution, and severity of the lesions. The absence of subretinal fluid, papillitis, and retinal vasculitis also can help differentiate APMPPE from other entities.
Discussion and Literature
First described by Gass in 1968, acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is a rare inflammatory condition within the white dot syndromes, a group of disorders characterized by multiple whitish-yellow inflammatory lesions located at the level of the outer retina, RPE, and choroid. It typically affects young, healthy individuals, presenting with a rapid onset of visual loss accompanied by central and paracentral scotomas. The disease is classically bilateral, with both eyes involved within a week, but may be asymmetrical. The incidence of APMPPE is approximately 0.15 per 100,000 people, mainly Caucasians, with no sex predilection.
The etiology of APMPPE is uncertain. It remains debated whether the choriocapillaris or the RPE is primarily involved. Evidence points to choriocapillaris hypoperfusion leading to ischemic injury of the RPE and photoreceptors, making it a choroidopathy rather than an epitheliopathy. Some even suggest APMPPE be renamed acute multifocal ischaemic choriocapillaritis (AMIC) and considered as part of the choriocapillaritis group.
About half of patients report a preceding febrile state, viral illness, or other systemic manifestation. Associations with multiple inflammatory conditions, infections, and vaccinations suggest that APMPPE is an immune-mediated phenomenon. A possible genetic predisposition has been proposed, with an increased risk associated with the HLA-B7 and HLA-DR2 haplotypes. Neurologic complications – including cerebral vasculitis, headache, aseptic meningitis, meningoencephalitis, sixth-nerve palsy, transient hearing loss, and cavernous sinus thrombosis – can occur during or after the ocular involvement.
Although the condition is considered self-limited, treatment is often indicated when foveal lesions are present to minimize retinal damage. Due to the rarity of the disease, there is no established consensus on the treatment of choice. Corticosteroids – oral, intravenous, or local – are commonly used, sometimes in combination with nonsteroidal anti-inflammatory drugs or immunosuppressants. Corticotherapy is tapered over several weeks, according to clinical response and imaging findings.
Visual prognosis is generally favorable – especially when compared with other white dot syndromes – with 87% of patients without foveal involvement recovering a visual acuity of 20/25 or better. Lesions normally resolve over several weeks, leaving various degrees of hyperpigmentation and RPE atrophy. Recurrences are rare, but some patients may experience persistent visual deficits.
Keep in mind
- APMPPE is a rare, bilateral, inflammatory condition associated with other immune triggers in young individuals.
- Multimodal image is the gold-standard in the diagnosis of APMPPE, with lesions characteristically larger and more numerous on ICG than on FFA.
- Anti-inflammatory therapy is helpful in preserving visual acuity in APMPPE; however, incomplete visual recovery may occur despite treatment.
References
- Quillen DA, Davis JB, Gottlieb JL, Blodi BA, Callanan DG, Chang TS & Equi RA (2004). The white dot syndromes. American journal of ophthalmology, 137(3), 538–550. https://doi.org/10.1016/j.ajo.2004.01.053
- Testi I, Vermeirsch S & Pavesio C (2021). Acute posterior multifocal placoid pigment epitheliopathy (APMPPE). Journal of ophthalmic inflammation and infection, 11(1), 31. https://doi.org/10.1186/s12348-021-00263-1
- Gallo B, Testi I, Grimaldi G, Rees A, Tucker W, Pavesio C & Westcott M (2025). Clinical and Imaging Features Aiding the Differentiation of Acute Posterior Multifocal Placoid Pigmented Epitheliopathy from Other Placoid Diseases. Ocular immunology and inflammation, 1–9. Advance online publication. https://doi.org/10.1080/09273948.2025.2518259
- Steiner S & Goldstein DA (2012). Imaging in the diagnosis and management of APMPPE. International ophthalmology clinics, 52(4), 211–219. https://doi.org/10.1097/IIO.0b013e318265d45a
- Heiferman MJ, Rahmani S, Jampol LM, Nesper PL, Skondra D, Kim LA & Fawzi AA (2017). ACUTE POSTERIOR MULTIFOCAL PLACOID PIGMENT EPITHELIOPATHY ON OPTICAL COHERENCE TOMOGRAPHY ANGIOGRAPHY. Retina (Philadelphia, Pa.), 37(11), 2084–2094. https://doi.org/10.1097/IAE.0000000000001487
- Oliveira MA, Simão J, Martins A & Farinha C (2020). Management of Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): Insights from Multimodal Imaging with OCTA. Case reports in ophthalmological medicine, 2020, 7049168. https://doi.org/10.1155/2020/7049168
- Fiore T, Iaccheri B, Androudi S, Papadaki TG, Anzaar F, Brazitikos P, D’Amico DJ & Foster CS (2009). Acute posterior multifocal placoid pigment epitheliopathy: outcome and visual prognosis. Retina (Philadelphia, Pa.), 29(7), 994–1001. https://doi.org/10.1097/IAE.0b013e3181a0bd15
- Xerri O, Salah S, Monnet D & Brézin AP (2018). Untreated Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): a case series. BMC ophthalmology, 18(1), 76. https://doi.org/10.1186/s12886-018-0744-z
- Papasavvas I, Mantovani A & Herbort CP Jr (2022). Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): A Comprehensive Approach and Case Series: Systemic Corticosteroid Therapy Is Necessary in a Large Proportion of Cases. Medicina (Kaunas, Lithuania), 58(8), 1070. https://doi.org/10.3390/medicina58081070
- Algahtani H, Alkhotani A & Shirah B (2016). Neurological Manifestations of Acute Posterior Multifocal Placoid Pigment Epitheliopathy. Journal of clinical neurology (Seoul, Korea), 12(4), 460–467. https://doi.org/10.3988/jcn.2016.12.4.460
