Presented by: Paris Tranos MD, PhD, FRCS, ICOphth & Jenny Papastergiopoulou MD


Edited by: Penelope Burle de Politis, MD

A 12-year-old boy was referred for investigation of peripapillary atrophy in one eye.
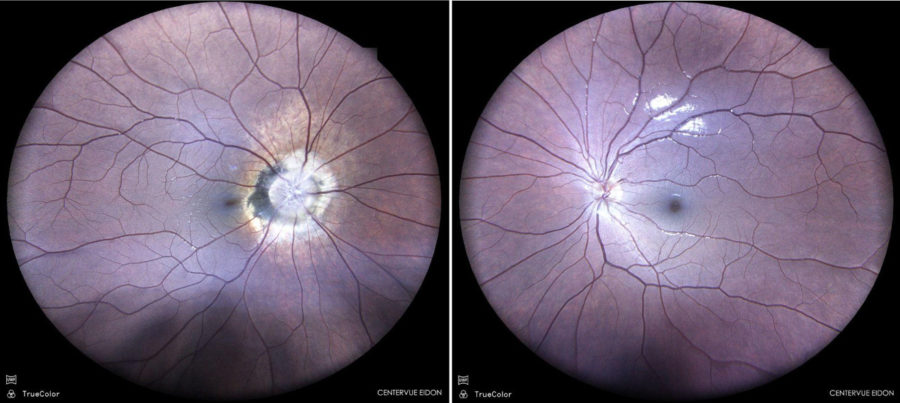
Figure 1: Color fundus photograph (EIDON true-color confocal scanner®) showing peripapillary atrophy and pigmentary changes in the right eye.
Case History
A 12-year-old Caucasian boy was referred for investigation of an accidentally found optic disc lesion in the right eye (RE). When questioned, he stated that his vision in the RE was somewhat worse than in the left eye (LE). There were no previous records of ocular disorders. He was a cystic fibrosis carrier. His family history was unremarkable. Upon ophthalmological examination, his corrected distance visual acuity (CDVA) was 10/10-1 in the RE and 10/10 in the LE. Refractometry was -0.75@40° in the RE and plano in the LE, and intraocular pressure was respectively 12 and 13 mmHg. There were no abnormalities under anterior segment biomicroscopy. At fundoscopy, LE was normal, while RE presented extensive peripapillary atrophy with pigmentary changes and abnormally rectified vessels. (Figure 1). There was no relative afferent pupillary defect (RAPD).
Fundus autofluorescence imaging of the RE displayed reduced autofluorescence at the peripapillary area (Figure 2).
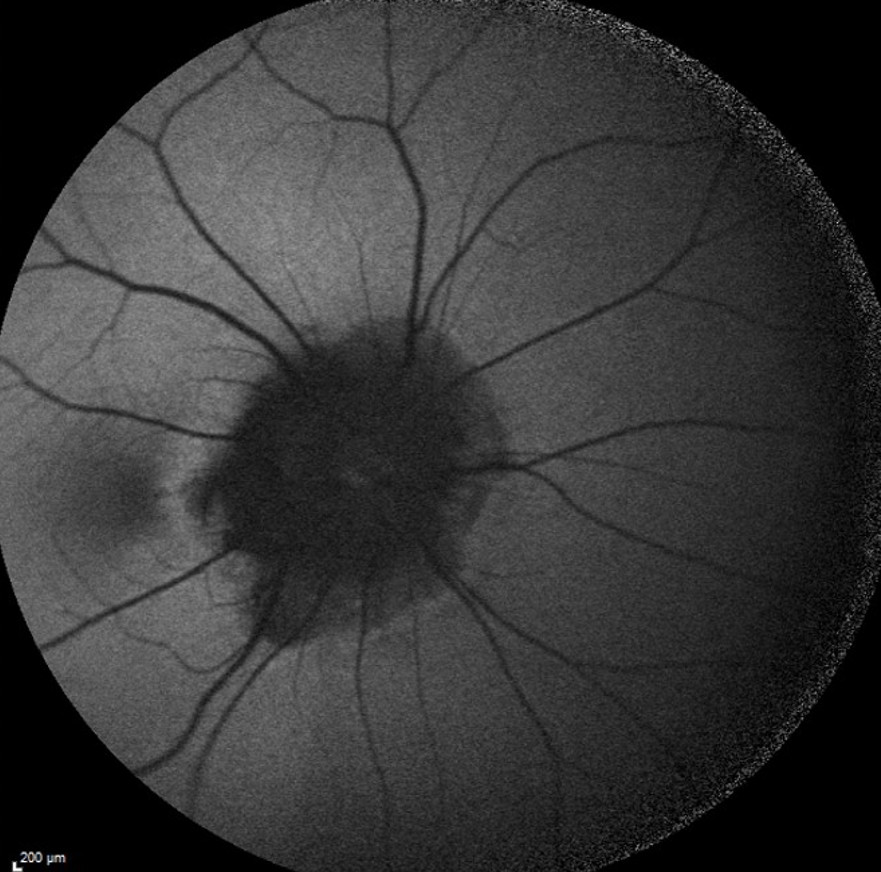
Figure 2: Fundus autofluorescence photograph of the RE (Heidelberg Engineering®) illustrating the extension of peripapillary atrophy and the vascular rectification.
Spectral domain optical coherence tomography (SD-OCT) of the RE evidentiated a deep central slope at the optic nerve head (Figure 3).
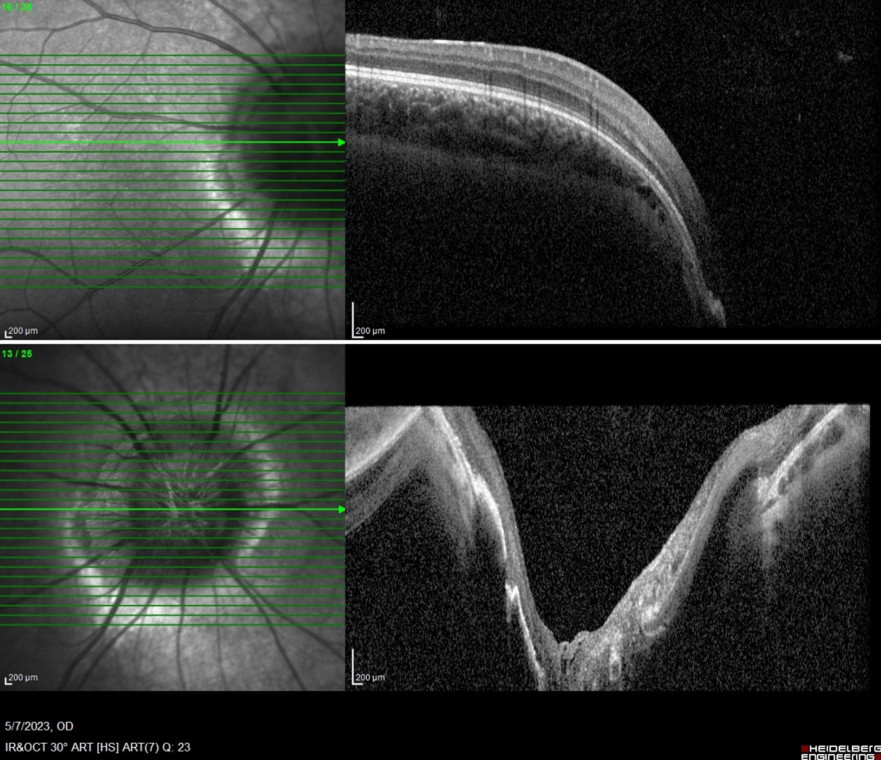
Figure 3: Spectral domain optical coherence tomography (SD-OCT, Heidelberg Engineering®) of the RE demonstrating a deep central pitting of the optic nerve head.
B-scan did not reveal any evidence of optic disc calcification (Figure 4).
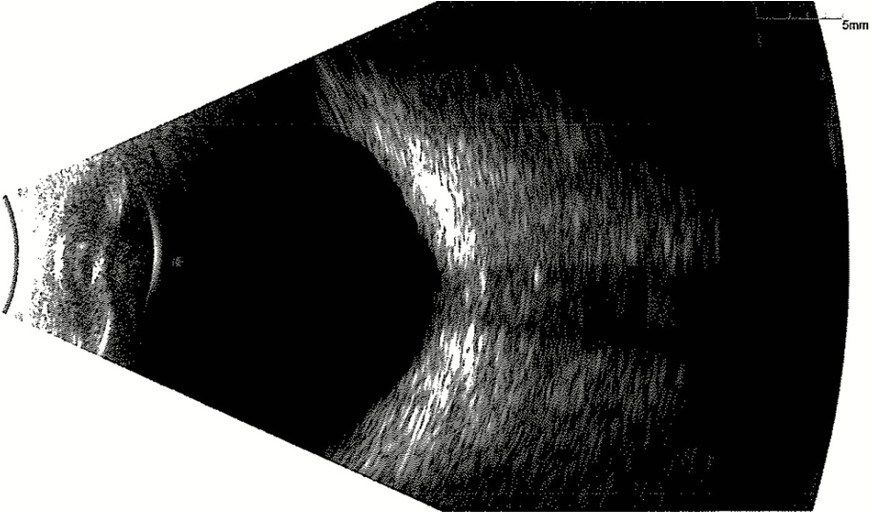
Figure 4: B-mode ultrasonography of the RE showing no evidence of calcification at the optic papilla.
Fundus reflectance imaging using different color filters showed a characteristic aspect of the optic nerve pitting, resembling the shape of the morning glory flower (Figure 5).
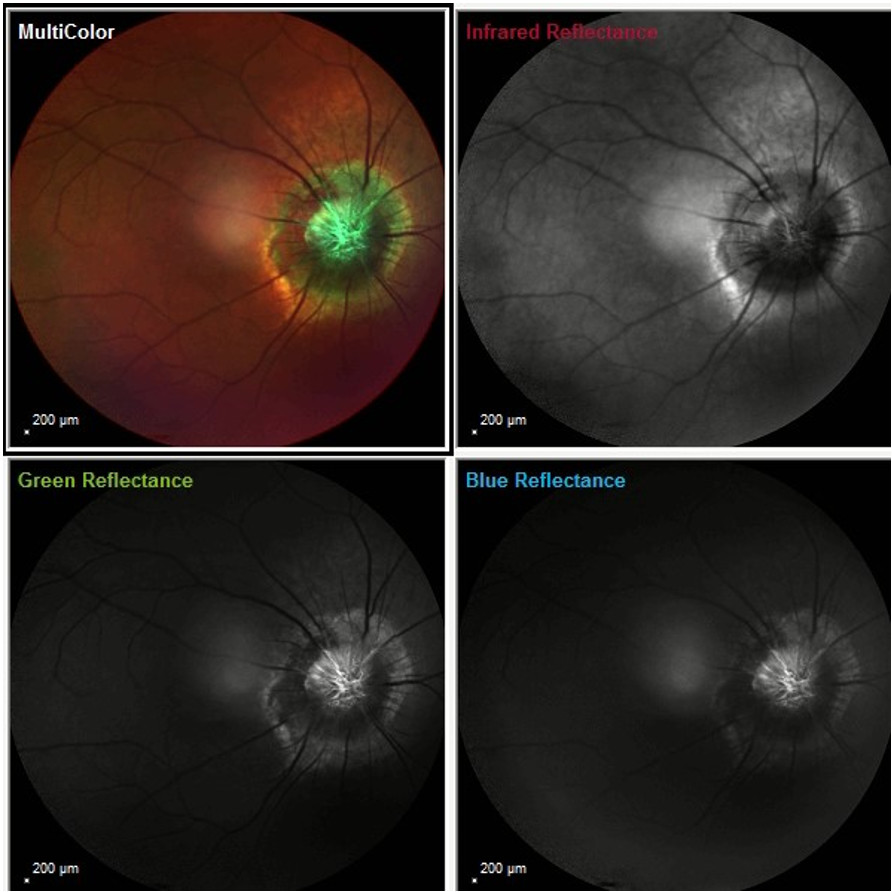
Figure 5: Infrared, blue, green, and multicolor reflectance photographs (Heidelberg Engineering®) of the RE featuring a characteristic flower-shaped pitting of the optic papilla. Notice the elevated rim outlining the optic nerve slope.
Additional History
Based on the clinical and multimodal image findings, the diagnosis of morning glory syndrome was established.
Brain imaging with magnetic resonance (MRI, MRA – magnetic resonance angiography) and/or computed tomography (CT, CTA – computed tomography angiography) was requested in order to rule out central nervous system (CNS) involvement, including basal encephalocele, Moyamoya syndrome (abnormal narrowing of cerebral arteries causing cerebral ischemia), and other structural or vascular abnormalities.
Considering the good visual acuity and absence of high-risk predisposing retinal lesions, conservative follow-up was the management of choice for the moment.
Differential Diagnosis of Morning Glory Syndrome
- optic disc coloboma
- CHARGE syndrome
- optic nerve head avulsion
- optic nerve aplasia
- optic disc pit
- advanced glaucomatous optic neuropathy
- combined retinal RPE hamartoma
- choroidal osteoma
- circumscribed choroidal hemangioma
- amelanotic choroidal nevus
The differentiation among the different types of optic papillary abnormalities is based on their morphological, histological, and vascular features. Attentive fundus examination supplemented with multimodal imaging methods is key for accurate diagnosing and management.
Discussion and Literature
Morning glory syndrome (MGS) or morning glory disc anomaly (MGDA) is a rare sporadic disorder characterized by a funnel-shaped optic nerve head. It was first described by Handmann in 1929, then by Kindler in 1970, who called it morning glory disc anomaly because of its resemblance to the morning glory flower. It is a primary mesenchymal and neuroectodermal malformation in which abnormal differentiation results in faulty closure of the posterior sclera and herniation of the retina and optic nerve head. The developmental disruption most likely occurs at 4-5 weeks of embryonic growth.
The reported prevalence of MGS is 2.6 to 3.6 per 100,000. A preference of 2:1 for the female gender has been reported and according to USA statistics it is less common in black people. It is mostly unilateral (85% of cases). Most of the cases present in early childhood with decreased vision – visual acuity usually ranging from 20/200 to counting fingers –, strabismus – esotropia or exotropia –, or leukocoria – less frequently. Bilateral cases normally present with nystagmus as well. Affected eyes are typically myopic and astigmatic. An afferent pupillary defect is usually present in unilateral cases.
Fundus evaluation of MGDA reveals an enlarged, orange or pink disc, within an excavated area. The peripapillary area is elevated, with an annular zone of pigmentary changes. The junction between the disc and the peripapillary area is often obscured. A whitish, fluffy, glial tissue is present in the center of the optic disc slope. The retinal blood vessels arise from the periphery of the excavation in a radial spoke, wheel-like pattern-disc and course radially towards the retinal periphery. They are abnormally straight and branch at acute angles. It is often difficult to differentiate an artery from a vein. Vascular sheathing may be present. The macula may be incorporated in the funnel-shaped excavated area, a finding known as macular capture. Retinoschisis may be present.
Some cases of MGDA have systemic associations such as Moyamoya syndrome, facial or central nervous system abnormalities, or renal coloboma syndrome. Magnetic resonance imaging of the skull and abdominal ultrasonography should be performed to discard involvement of other organs. Computerized tomography (CT) of the orbits and brain can reveal intraocular calcifications and other abnormalities. Early diagnosis is crucial to avoid complications, mainly retinal detachment (RD), either tractional — due to subretinal neovascularization —, rhegmatogenous or serous (with or without retinal breaks). It is hypothesized that small breaks can form due to the contractile nature of the glial tissue, allowing for the accumulation of subretinal fluid. Eco- and tomographic findings point to the existence of an abnormal communication between the subretinal and subarachnoid or vitreous compartments, another explanation for the physiopathology of RD in MGS.
The severity of MGDA and comorbidities in an individual patient may not be revealed through routine ophthalmological examination, which calls for attention to associated risks. This is especially important as most patients are diagnosed at an age where intervention still can be highly meaningful. Refractive errors should be corrected when present to improve visual acuity and prevent amblyopia. Juxtapapillary laser photocoagulation can be used both prophylactically or supplementarily to vitrectomy in the prevention and management of MGS-associated RD. Recurrence and complications of the surgical interventions must be taken into consideration. Ecographic parameters such as cavitary depth, size of excavation and axial length can be used to assess the risk of RD and poor vision.
Keep in mind
- Morning glory syndrome is a rare, usually unilateral malformation of the optic papilla, and sometimes of other organs and systems. Neuroimaging is recommended to rule out CNS involvement.
- Morning glory disc anomaly classically presents during childhood, with decreased vision in the affected eye and strabismus.
- The most frequent complication of MGDA is RD; therefore, extensive search for predisposing lesions is crucial to prevent vision loss.
References
- Gupta, A., Singh, P., & Tripathy, K. (2023). Morning Glory Syndrome. In StatPearls. StatPearls Publishing.
- Kindler P. (1970). Morning glory syndrome: unusual congenital optic disk anomaly. American journal of ophthalmology, 69(3), 376–384. https://doi.org/10.1016/0002-9394(70)92269-5
- Pollock S. (1987). The morning glory disc anomaly: contractile movement, classification, and embryogenesis. Documenta ophthalmologica. Advances in ophthalmology, 65(4), 439–460. https://doi.org/10.1007/BF00143047
- Dedhia, C. J., Gogri, P. Y., & Rani, P. K. (2016). Rare bilateral presentation of morning glory disc anomaly. BMJ case reports, 2016, bcr2016215846. https://doi.org/10.1136/bcr-2016-215846
- Ceynowa, D. J., Wickström, R., Olsson, M., Ek, U., Eriksson, U., Wiberg, M. K., & Fahnehjelm, K. T. (2015). Morning glory disc anomaly in childhood – a population-based study. Acta ophthalmologica, 93(7), 626–634. https://doi.org/10.1111/aos.12778
- Zou, Y., She, K., Hu, Y., Ren, J., Fei, P., Xu, Y., Peng, J., & Zhao, P. (2022). Clinical and Echographic Features of Morning Glory Disc Anomaly in Children: A Retrospective Study of 249 Chinese Patients. Frontiers in medicine, 8, 800623. https://doi.org/10.3389/fmed.2021.800623
- Cennamo, G., de Crecchio, G., Iaccarino, G., Forte, R., & Cennamo, G. (2010). Evaluation of morning glory syndrome with spectral optical coherence tomography and echography. Ophthalmology, 117(6), 1269–1273. https://doi.org/10.1016/j.ophtha.2009.10.045
- Harasymowycz, P., Chevrette, L., Décarie, J. C., Hanna, N., Aroichane, M., Jacob, J. L., Milot, J., & Homsy, M. (2005). Morning glory syndrome: clinical, computerized tomographic, and ultrasonographic findings. Journal of pediatric ophthalmology and strabismus, 42(5), 290–295. https://doi.org/10.3928/0191-3913-20050901-11
- Zou, Y. H., She, K. Q., Ren, J. N., Liang, T. Y., Fei, P., Xu, Y., Li, J., Zhang, X., Peng, J., & Zhao, P. Q. (2022). Prophylactic juxtapapillary laser photocoagulation in pediatric morning glory syndrome. International journal of ophthalmology, 15(5), 766–772. https://doi.org/10.18240/ijo.2022.05.12
- Etheridge, T., Oakey, Z., & Altaweel, M. M. (2021). Management of Retinal Detachment Associated with Morning Glory Disc Syndrome. Case reports in ophthalmology, 12(2), 457–463. https://doi.org/10.1159/000516205
