Presented by: Chrysa Koutsiouki MD

Edited by: Penelope Burle de Politis, MD

A 30-year-old myope woman presented with floaters and progressive visual loss in the left eye over the course of 3 months.
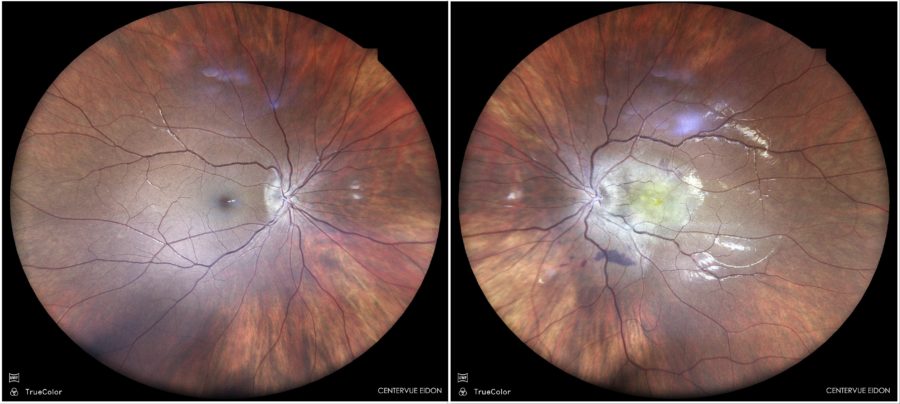
Figure 1: Fundus color photograph (iCare confocal high-resolution ultra-widefield Truecolor Centervue EIDON®) showing a yellowish-white lesion at the posterior pole of the left eye involving the macula.
Case History
A 30-year-old Caucasian woman presented with progressively decreased vision and floaters in the left eye (LE) noticed three months earlier. She had 3.00D myopia in both eyes (BE). Her past medical history was limited to well-controlled hypothyroidism from Hashimoto’s disease. Her family medical history was negative for ophthalmic diseases. Upon examination, her corrected distance visual acuity (CDVA) was 10/10 in the right eye (RE) and hand movement (HM) in the LE. The anterior ocular segment was normal in BE. Intraocular pressure (IOP) was within the normal range bilaterally. Fundus examination of the LE showed a poorly defined central yellowish-white lesion, with macular involvement and significant retinal folds. There were no abnormalities in the RE (Figure 1). Further investigation using multimodal imaging revealed intraretinal lesions in the macular area of the LE, with accompanying edema and neovascularization (Figures 2 to 6).
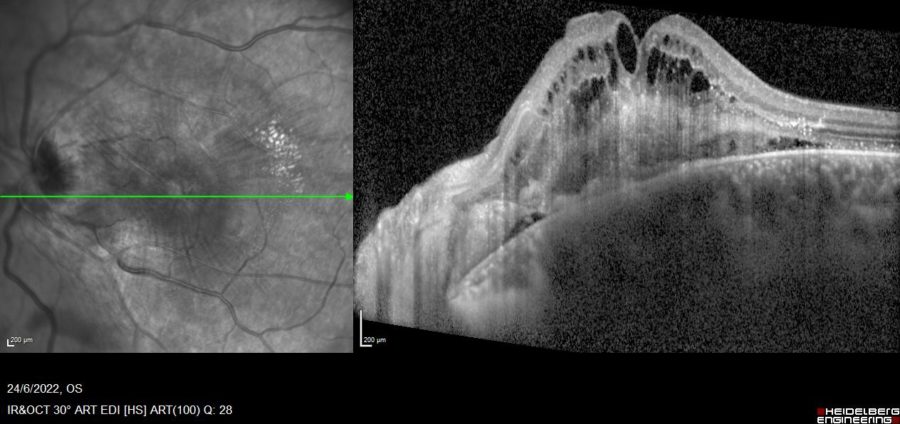
Figure 2: Enhanced depth mode of spectral-domain optical coherence tomography (EDI SD-OCT) of the LE (Spectralis, Heidelberg Engineering®) displaying increased retinal thickness, a hyperreflective material in the inner retina with loss of retinal architecture, and cystoid macular edema (CMO).
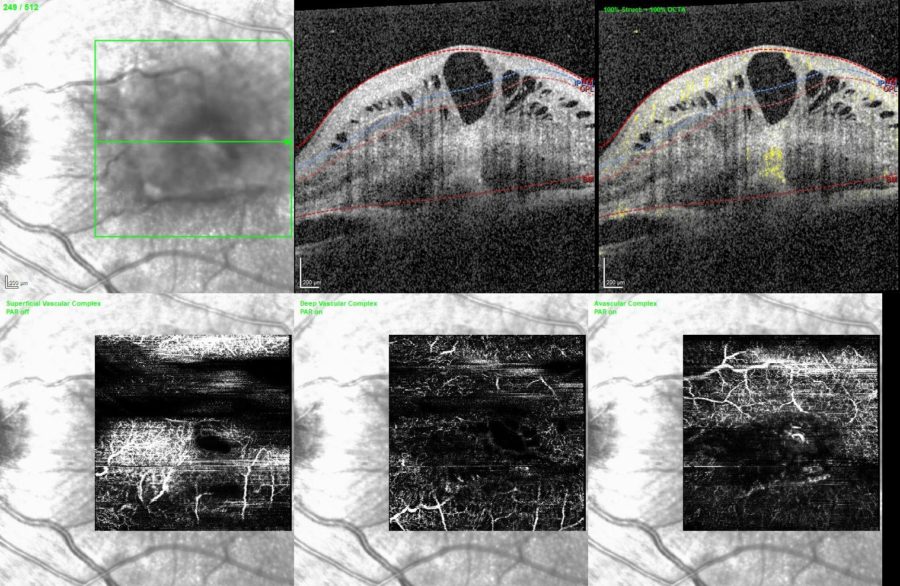
Figure 3: Optical coherence tomography angiography (OCT-A) of the LE (Spectralis, Heidelberg Engineering®) showing abnormal retinal vessels in the superficial (SCP) and deep retinal capillary plexuses (DCP), with evidence of flow.
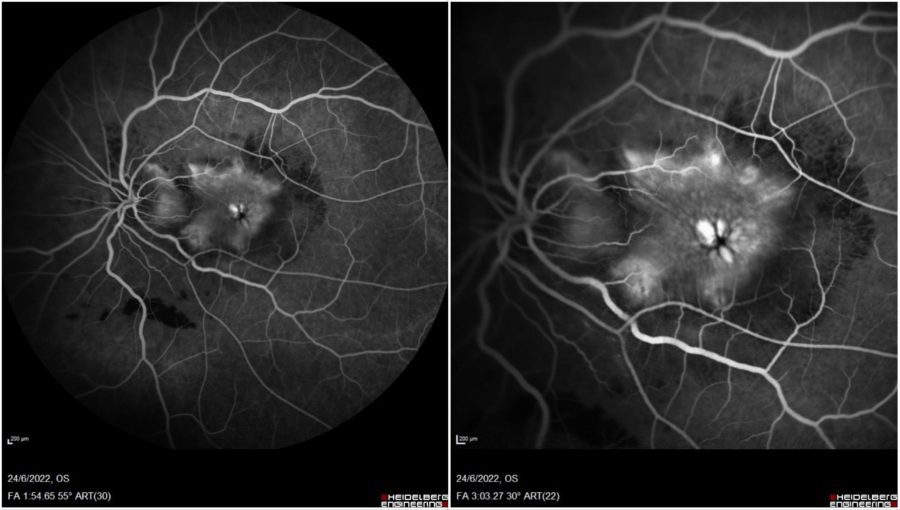
Figure 4: Fundus fluorescein angiography (FFA) of the LE (Spectralis, Heidelberg Engineering®). Early frame (right): hypofluorescence from retinal fold masking, dilated perifoveal capillaries, progressively increasing leakage. Late frame (left): cystoid macular edema.
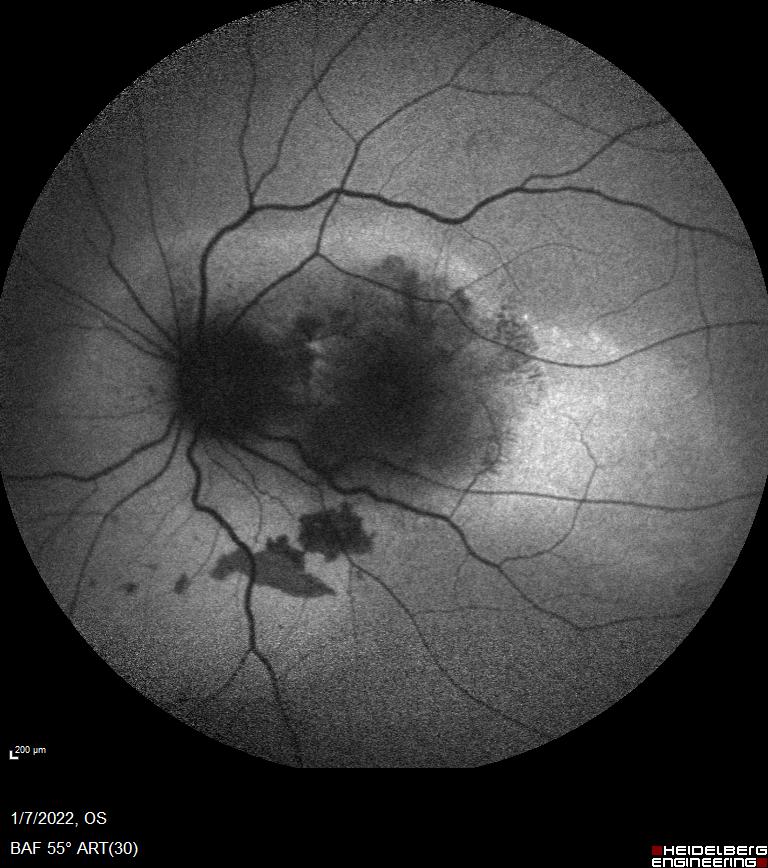
Figure 5: Blue-light fundus autofluorescence (BAF) of the LE (Spectralis, Heidelberg Engineering®) showing the hypoautofluorescence of the lesions with mild hyperautofluorescence in the nasal area.
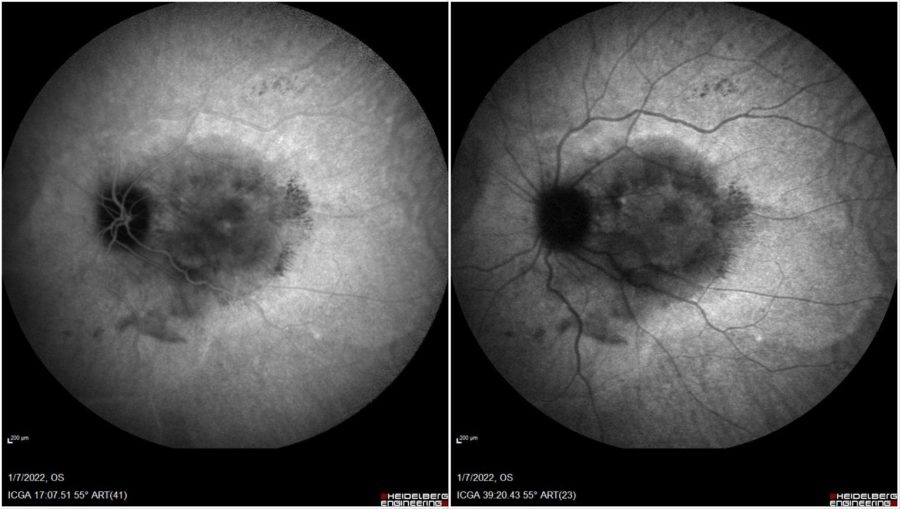
Figure 6: Indocyanine-green angiography (ICG-A) of the LE (Spectralis, Heidelberg Engineering®). Early frame (right): hypofluorescence, along with retinal vessels crossing the foveal area. Late frame (left): hyperfluorescence of the abnormal vessels.
Additional History
Based on the clinical and image findings, the diagnosis of combined hamartoma of the retina and retinal pigment epithelium was established. Considering the acute-onset blurred vision and the presence of intraretinal fluid and neovascularization, intravitreal anti-VEGF was indicated. The prognostic perspectives were discussed with the patient and the injection was carried out. Follow-up imaging showed regression of the exudative component and improvement of retinal thickening (Figure 7), though no improvement in visual acuity could be noticed.
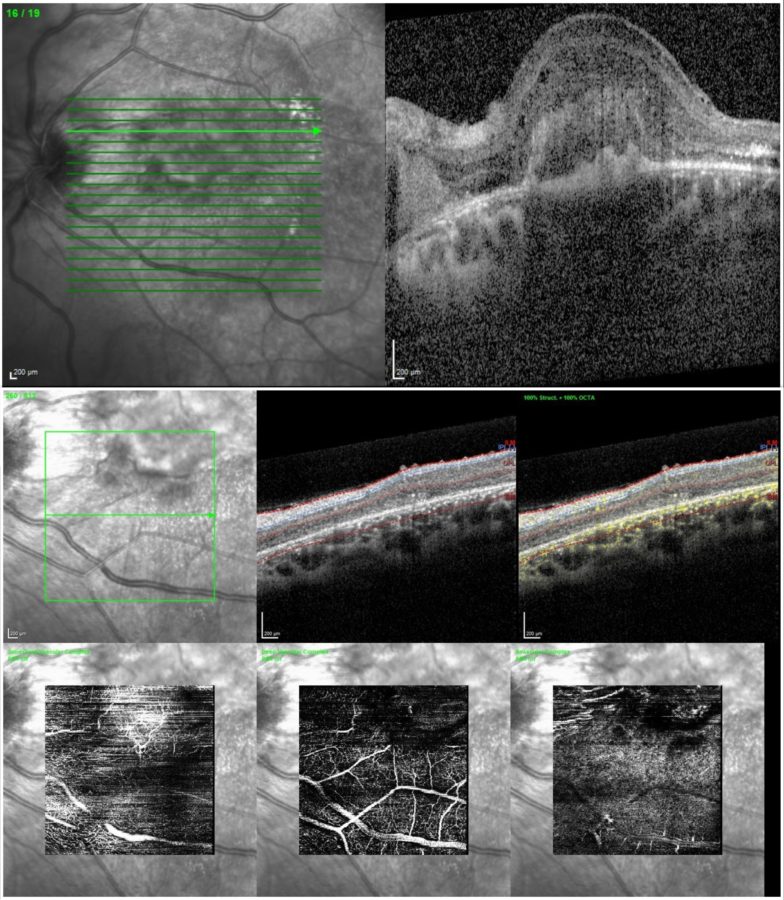
Figure 7: EDI SD-OCT (top) and OCT-A (bottom) of the LE at 6 weeks post-Avastin® (bevacizumab) injection, showing significant resolution of the cystoid macular edema, although with persistence of the hyper-reflective material.
Differential Diagnosis of Combined Hamartoma of the Retina and RPE
- choroidal melanoma
- choroidal nevus
- primary or secondary retinal gliosis
- adenoma or adenocarcinoma of RPE
- melanocytoma
- morning glory anomaly
- retinoblastoma
- toxocariasis
- astrocytoma
- hemangioma
Because combined hamartoma of the retina and retinal pigment epithelium can resemble other posterior pole lesions, accurate diagnosis using multimodal imaging technology is of ultimate importance for decision-making in case management.
Discussion and Literature
The term hamartoma derives from the Greek “hamartia,” meaning error. Hamartomas are benign, slow overgrowths that result from the abnormal proliferation of cells in regions where they are normally found. They occur in different parts of the body and most cases are asymptomatic and detected incidentally. Hamartomas can occur isolatedly or in association with a genetic syndrome. The retinal and optic disc hamartomas types are: astrocytic hamartoma, congenital hypertrophy of the retinal pigment epithelium (CHRPE), congenital simple hamartoma of the retinal pigment epithelium (CSHRPE), combined hamartoma of the retina and retinal pigment epithelium (CHR-RPE), retinal hemangioblastoma (retinal capillary hemangioma), and retinal cavernous hemangioma. For CHR-RPE, the possible systemic associations include neurofibromatosis types 1 and 2, Gorlin-Goltz syndrome, Poland anomaly, branchio-oculo-facial syndrome, brachio-oto-renal syndrome, and juvenile nasopharyngeal angiofibroma.
CHR-RPE is a rare lesion, usually presenting between the ages of 1 and 74 years (mean age 23 years). It is generally congenital and nonhereditary, and its etiology is uncertain. Males and females were equally affected. Caucasian race appears to predominate. The lesion is generally unilateral and most cases are sporadic. Bilateral lesions should raise suspicion of neurofibromatosis. Histologically, CHR-RPE is composed of a mixture of pigment epithelial cells, proliferating blood vessels, and glial tissue, located mostly in the sensory retina or optic disc tissue. It is believed to originate from the inner retina and progress towards the outer retina over time.
Visual acuity is variable, depending on lesion size and location and amount of retinal traction. Most patients with CHR-RPE that come to clinical attention are young children found to have poor vision on school examinations. The patients commonly present with a painless decrease in VA, strabismus, or with nonspecific ocular irritation. Less common presenting symptoms include floaters, ocular pain, and leukocoria. Tumors located in the macular area can lead to strabismus and amblyopia. Foveal ectopia secondary to traction by a peripheral lesion can also be a cause of amblyopia.
The CHR-RPE lesion has characteristic but variable clinical features. The most typical finding is an ill-defined retinal mass with tortuous or straightened retinal blood vessels. The tumor can be dusky brown, green, yellow, gray, or orange. The classic location is on or adjacent to the optic disc but other areas of the fundus may be involved. It can vary widely in size, ranging from a small 1-mm to a mass lesion larger than 10 mm in diameter. While young patients exhibit mainly inner retinal involvement, full-thickness retinal involvement is often seen in older patients. An increase in retinal thickness is more common in macular lesions than in other locations. Peripheral lesions can cause a “dragged disc” appearance due to retinal traction.
CHR-RPE was generally classified into three groups according to location: peripapillary, macular, and peripheral. However, a recent classification system has been proposed, based on tumor’s location (posterior, mid-periphery, or far periphery), characteristics (presence of traction, retinoschisis, or retinal detachment), and OCT findings (epiretinal, partial retinal, or full-thickness retina/RPE involvement). According to the stage, management options can range from complete ophthalmologic evaluation at least every 6 months to surgical intervention.
On OCT, CHR-RPE exhibits an irregular lesion with vitreoretinal traction into a “sawtooth” or “folded” pattern that replaces full-thickness retinal tissue (mini-peak) and “omega sign” (maxi-peak). OCT-A demonstrates retinal vascular alterations and a “filigree” pattern in the intratumoral vessels. On FFA, the lesion is hyperfluorescent with early hypoflourescence, and there are markedly abnormal vessels in the mass, with gradual late staining of the lesion. Peripapillary pigmented lesions exhibit hyperautofluorescence on FAF. Indocyanine green angiography shows mild patchy hyperfluorescence in the late phase. Ultrasonography may help rule out other conditions with similar appearance. The usual ultrasonographic aspect is a plaque-like, slightly raised, acoustically solid lesion with moderate to high internal reflectivity and no extrascleral extension. If neurofibromatosis type II is suspected, further work-up is warranted.
Patients with CHR-RPE can suffer from progressive visual loss due to tumor growth or complications associated with the tumor, which include amblyopia, formation of ERM, retinal holes, retinoschisis, CNV, retinal neovascularization, vitreous hemorrhage, and retinal detachment. There is no highly effective method for treating CHR-RPE. When CHR-RPE presents at a young age, amblyopia prevention is paramount. Surgery for associated ERMs is still a subject of debate since visual acuity may not improve despite membrane removal. Also, it may not be possible to remove the whole ERM without damaging the retina and is not feasible if the lesion involves full thickness of the retina. Laser photocoagulation can be employed for neovascular membranes. Intravitreal anti-VEGF injection can be used to treat choroidal neovascularization.
Keep in mind
- CHR-RPE usually presents early in life and is found incidentally at any age.
- Posterior location of CHR-RPE is associated with lower visual acuity but complications can occur in peripheral lesions as well.
- Management of CHR-RPE is case-based and aimed at preserving visual acuity.
References
- Shields JA & Shields CL (2017). Tumors and Related Lesions of the Pigmented Epithelium. Asia-Pacific journal of ophthalmology (Philadelphia, Pa.), 6(2), 215–223. https://doi.org/10.22608/APO.201705
- Mirzayev I & Gündüz AK (2022). Hamartomas of the Retina and Optic Disc. Turkish journal of ophthalmology, 52(6), 421–431. https://doi.org/10.4274/tjo.galenos.2022.25979
- Schachat AP, Shields JA, Fine SL, Sanborn GE, Weingeist TA, Valenzuela RE & Brucker AJ (1984). Combined hamartomas of the retina and retinal pigment epithelium. Ophthalmology, 91(12), 1609–1615. https://doi.org/10.1016/s0161-6420(84)34094-5
- Dedania VS, Ozgonul C, Zacks DN & Besirli CG (2018). NOVEL CLASSIFICATION SYSTEM FOR COMBINED HAMARTOMA OF THE RETINA AND RETINAL PIGMENT EPITHELIUM. Retina (Philadelphia, Pa.), 38(1), 12–19. https://doi.org/10.1097/IAE.0000000000001499
- Ren Q, Han N, Zhang R, Chen RF & Yu P (2023). Combined hamartoma of the retina and retinal pigment epithelium: A case report. World journal of clinical cases, 11(8), 1788–1793. https://doi.org/10.12998/wjcc.v11.i8.1788
- Ledesma-Gil G, Essilfie J, Gupta R, Fung AT, Lupidi M, Pappuru RR, Nayak S, Sahoo NK, Kaliki S, Yannuzzi LA, Reid K, Lim L, Sacconi R, Dave V, Singh SR, Ayachit A, Gabrielle PH, Cai S, Lima LH, Querques G et al. (2021). Presumed Natural History of Combined Hamartoma of the Retina and Retinal Pigment Epithelium. Ophthalmology. Retina, 5(11), 1156–1163. https://doi.org/10.1016/j.oret.2021.01.011
- Gupta R, Fung AT, Lupidi M, Pappuru RR, Nayak S, Sahoo NK, Kaliki S, Yannuzzi L, Reid K, Lim L, Sacconi R, Dave V, Singh SR, Ayachit A, Gabrielle PH, Cai S, Lima LH, Querques G, Arevalo JF, Freund KB et al. (2019). Peripapillary Versus Macular Combined Hamartoma of the Retina and Retinal Pigment Epithelium: Imaging Characteristics. American journal of ophthalmology, 200, 263–269. https://doi.org/10.1016/j.ajo.2019.01.016
- Kaprinis K, Bobat H & De Salvo G (2018). MultiColorTM imaging in combined hamartoma of the retina and retinal pigment epithelium. Eye (London, England), 32(9), 1478–1482. https://doi.org/10.1038/s41433-018-0123-2
- Shields CL, Thangappan A, Hartzell K, Valente P, Pirondini C & Shields JA (2008). Combined hamartoma of the retina and retinal pigment epithelium in 77 consecutive patients visual outcome based on macular versus extramacular tumor location. Ophthalmology, 115(12), 2246–2252.e3. https://doi.org/10.1016/j.ophtha.2008.08.008
- Inoue M, Noda K, Ishida S, Yamaguchi T, Nagai N, Shinoda K, Shinoda H & Oguchi Y (2004). Successful treatment of subfoveal choroidal neovascularization associated with combined hamartoma of the retina and retinal pigment epithelium. American journal of ophthalmology, 138(1), 155–156. https://doi.org/10.1016/j.ajo.2004.02.020
