Presented by: Dimitris Sakellaris MD

Edited by: Penelope Burle de Politis, MD

A 26-year-old woman presented with longstanding, slowly progressive bilateral low visual acuity and recurrent ocular irritation and foreign body sensation.
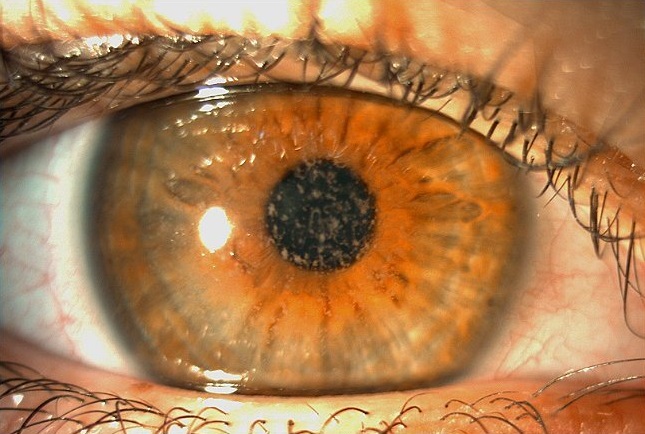
Figure 1: Slit-lamp photograph of the right eye displaying heterogeneous stromal deposits interspersed by areas of normal corneal stroma.
Case History
A 26-year-old Caucasian woman presented with a history of slowly progressively low visual acuity and frequent ocular surface irritation and foreign body sensation in both eyes since childhood. She had been wearing glasses for 20 years and would like to know if her eyes were suitable for laser correction. She denied any previous contact lens usage and was not on any medication. Upon ophthalmological examination, (autorefractometer measurements) (were -0.50@165° in the right eye (RE) and -1.00 -1.00@10° in the left eye (LE), with a distance corrected visual acuity (DCVA) of 5/10-2 bilaterally. Fundoscopy was unremarkable and intraocular pressure (IOP) was 13 mmHg in both eyes. Pupils were normally reactive to light and there were no extraocular muscle imbalances. Biomicroscopy revealed heterogeneous anterior stromal corneal deposits permeated with areas of normal corneal stroma in both eyes (Figure 1).
Preservative-free artificial tears were prescribed and started on a regular basis. Symptomatic relief was achieved after a few days of treatment, with a slight subjective improvement in vision as well. Surgical treatment options were discussed but put on hold, given the current clinical presentation and their risks and benefits.
Additional History
The patient’s father had a history of some sort of keratopathy and cataract in his RE, having undergone a scraping procedure for diagnosis a few years before. He had also been told he could need eye drops for ocular hypertension, which he actually never used. He was 58 years-old and presented a refraction error of +3.00 -0.50@15° in the LE, with a DCVA of 8/10+2. In the RE, refractometry was hardly feasible and the best achievable DCVA was 4/10 with a correction of +2.00 -2.50@180°. IOP was 21 and 18 in the RE and LE respectively, and there were no fundus abnormalities. Under biomicroscopic examination, he was found to have a mild nuclear sclerosis and a corneal aspect identical to his daughter’s, more pronounced in his RE (Figure 2).
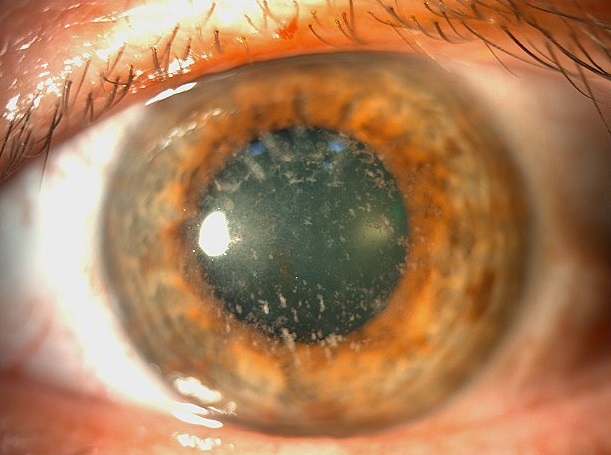
Figure 2: Slit-lamp photograph of the father’s right eye displaying anterior stromal deposits in a coarser pattern but similar to that seen in his daughter’s cornea.
The ocular history and clinical findings, added to the spontaneous occurrence in direct family members, led to the diagnosis of a corneal dystrophy. The mixed nature of the stromal deposits, both granular and lattice in appearance, were characteristic of Granular Corneal Dystrophy Type 2 (GCD2 or Avellino type).
Differential Diagnosis of Granular Corneal Dystrophy Type 2 (GCD2)
- cystinosis
- recurrent corneal erosion
- keratoconjunctivitis sicca
- adenoviral keratitis
- monoclonal gammopathy
The main differential diagnosis of GCD2 is with other forms of corneal dystrophies. Aside from careful slit-lamp examination, biochemical analysis of scraped-off deposits and genetic testing can help in diagnosis.
Discussion and Literature
Corneal dystrophies (CD) are a heterogenous group of bilateral, genetically determined, non-inflammatory diseases restricted to the cornea. Clinically, CD can be divided into three groups based on the sole or predominant anatomical location of the abnormalities. Some affect primarily the corneal epithelium and its basement membrane, or Bowman’s layer and the anterior corneal stroma (anterior CD), others the mid and posterior corneal stroma (stromal CD), or Descemet’s membrane and the corneal endothelium (posterior CD).
Most CD present with variably shaped corneal opacities in a clear or cloudy cornea and affect visual acuity to different degrees. The inheritance of CD may be autosomal dominant, autosomal recessive or X-linked recessive. The available molecular tests for their precise diagnosis are based on the knowledge of the genetic mutation responsible for each corneal dystrophy. A corneal dystrophy should be suspected when corneal transparency is lost or when corneal opacities occur spontaneously, particularly in both corneas, and especially in the presence of a positive family history or in the offspring of consanguineous parents.
Granular corneal dystrophies (GCD) belong to the stromal CD. Subtle differences in the clinical appearance of the corneal opacities characterize two types of GCD: GCD type I (GCD1, Groenouw type) and GCD type II (GCD2, Avellino type). GCD2 tends to have fewer corneal deposits than GCD1, and the corneal deposits in GCD2 sometimes resemble a combination of GCD and lattice CD, featuring both granular and amyloid deposits. GCD1 seems to be most prevalent in Europe, while GCD2 is more common in Japan, Korea and the USA. The ancestry of some families with GCD2 have been traced to the Avellino district of Italy – hence the synonym Avellino corneal dystrophy.
GCD2 is an autosomal dominant disorder and results from the mutation p.Arg124His (arginine to histidine substitution at codon 124) in the transforming growth factor β-induced (TGFBI) gene, also referred to as TGFBI p.R124H. Patients with GCD2 have different ages of onset and progression rates, depending on whether the p.R124H variant is heterozygous or homozygous. In patients with the homozygous p.R124H variant, corneal deposits appear as early as 3 years of age, and visual loss begins in childhood. On the other hand, heterozygous patients have a highly variable phenotype due to incomplete penetrance even in the same family. They are usually diagnosed as teenagers or young adults and progress slowly.
The severity of opacity in CD might not always correlate to visual function. Recent technologies such as anterior segment optical coherence tomography (AS-OCT) and wavefront-based aberrometry has allowed for better understanding of how CD affect vision. It has been demonstrated that the stromal opacities of CD induce not only scatter but also morphometric changes in the anterior and posterior corneal surfaces that can cause high-order aberrations (coma, trefoil, spherical aberration etc.). In GCD2, those are lesser than in other forms of CD such as lattice and macular, having different repercussions on patients’ visual acuity.
To treat the opacities in GCD2, phototherapeutic keratectomy (PTK), lamellar keratoplasty, deep lamellar keratoplasty, and penetrating keratoplasty (PKP) have been used. However, recurrence is still an unsolved problem, because corneal trauma activates TGFBI, the main component of the corneal opacities. In fact, several studies have reported an exacerbation of GCD2 after corneal refractive surgery such as photorefractive keratectomy (PRK), laser in situ keratomileusis (LASIK), and laser epithelial keratomileusis (LASEK). For this reason, TGFBI gene screening is advisable for candidates with a family history of corneal disease or granular opacities in corneal stroma before refractive surgery.
The pathogenesis of GCD2 lies in lysosomal dysfunction and autophagy of corneal fibroblasts. In GCD2 corneal fibroblasts, mutant-TGFBIp accumulates in the lysosomal compartments due to defective autophagy. Transcription factor EB (TFEB) plays an essential role in cellular homeostasis, modulating lysosomal function and autophagy. New research sheds light on TFEB as a potential therapeutic target for maintaining the physiological function of corneal fibroblasts. Apparently, exogenous TFEB expression improves mutant-TGFBIp clearance and lysosomal abnormalities in GCD2 cells. Preclinical studies to evaluate the possible consequences of TFEB overexpression or activation are still pending.
Keep in mind
- A corneal dystrophy should always be suspected when corneal opacities occur spontaneously, particularly if bilaterally, and especially in the presence of a positive family history.
- GCD2 phenotypes may vary widely, even in the same family, depending on heterozygosity or homozygosity and on complete or incomplete penetrance of the mutant gene.
- Pros and cons of therapeutic procedures for GCD2 must be weighed against the visual deficit presented by each patient and its effects on their quality of life.
References
- Klintworth GK (2009). Corneal dystrophies. Orphanet journal of rare diseases, 4, 7. https://doi.org/10.1186/1750-1172-4-7
- Park JE, Yun SA, Roh EY, Yoon JH, Shin S & Ki CS (2021). Prevalence of granular corneal dystrophy type 2-related TGFBI p.R124H variant in a South Korean population. Molecular vision, 27, 283–287. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8116257/
- Han KE, Kim TI, Chung WS, Choi SI, Kim BY & Kim EK (2010). Clinical findings and treatments of granular corneal dystrophy type 2 (avellino corneal dystrophy): a review of the literature. Eye & contact lens, 36(5), 296–299. https://doi.org/10.1097/ICL.0b013e3181ef0da0
- Han KE, Choi SI, Chung WS, Jung SH, Katsanis N, Kim TI & Kim EK (2012). Extremely varied phenotypes in granular corneal dystrophy type 2 heterozygotes. Molecular vision, 18, 1755–1762. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3398492/
- Abazi Z, Magarasevic L, Grubisa I & Risovic D (2013). Individual phenotypic variances in a family with Avellino corneal dystrophy. BMC ophthalmology, 13, 30. https://doi.org/10.1186/1471-2415-13-30
- Yagi-Yaguchi Y, Yamaguchi T, Okuyama Y, Satake Y, Tsubota K & Shimazaki J (2016). Corneal Higher Order Aberrations in Granular, Lattice and Macular Corneal Dystrophies. PloS one, 11(8), e0161075. https://doi.org/10.1371/journal.pone.0161075
- Abe Y, Omoto T, Kitamoto K, Toyono T, Yoshida J, Asaoka R, Yamagami S, Miyai T & Usui T (2022). Corneal irregularity and visual function using anterior segment optical coherence tomography in TGFBI corneal dystrophy. Scientific reports, 12(1), 13759. https://doi.org/10.1038/s41598-022-17738-3
- Rana RS, Bajracharya L & Gurung R (2021). Recurrence of Avellino Corneal Dystrophy Following Penetrating Keratoplasty: A Case Report. JNMA; journal of the Nepal Medical Association, 59(236), 406–408. https://doi.org/10.31729/jnma.5726
- Jiang X & Zhang H (2021). Deterioration of Avellino corneal dystrophy in a Chinese family after LASIK. International journal of ophthalmology, 14(6), 795–799. https://doi.org/10.18240/ijo.2021.06.02
- Choi SI, Woo JH & Kim EK (2020). Lysosomal dysfunction of corneal fibroblasts underlies the pathogenesis of Granular Corneal Dystrophy Type 2 and can be rescued by TFEB. Journal of cellular and molecular medicine, 24(18), 10343–10355. https://doi.org/10.1111/jcmm.15646
