Presented by: Georgios Sidiropoulos MD, FEBOphth

Edited by: Penelope de Politis, MD

A 33 year-old woman presented asymptomatic corneal flecks in a routine eye exam.
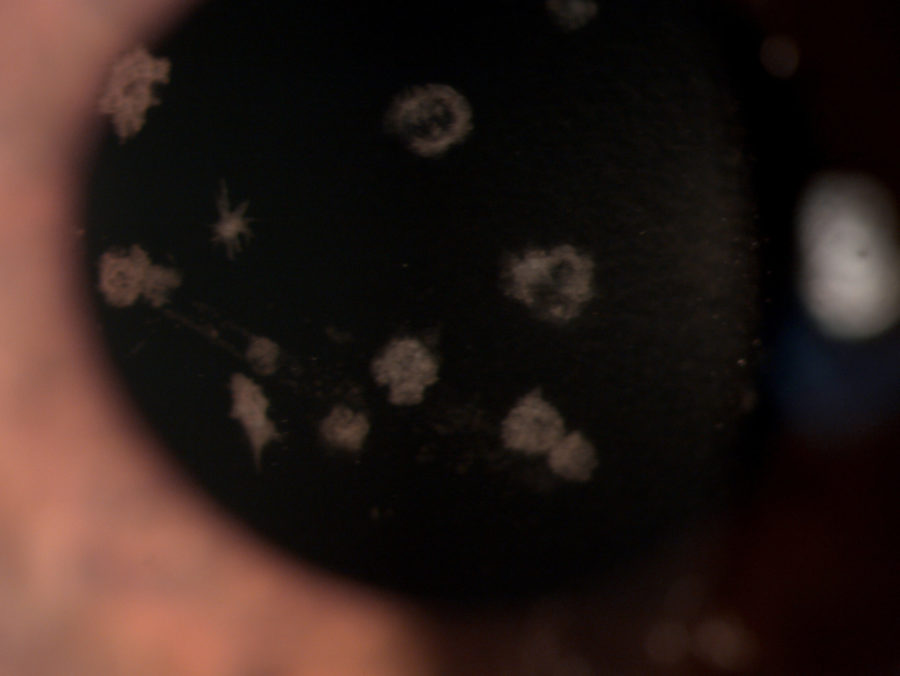
Figure 1: Microscope photograph with broad beam.
Case History
A 33 year-old Caucasian woman presented bilateral corneal opacities during routine ophthalmic examination. Her only complaint was astenopia related to prolonged computer use. On examination, uncorrected visual acuity (UCVA) was 12/10 bilaterally. Objective refraction was -0,50@75∘ on the right and plano on the left. Intraocular pressure and fundoscopy were normal on both eyes. Biomicroscopy revealed calm eyes displaying whitish spiky granules at stroma level of both corneas, best evidentiated upon retroillumination through the pupillary area (Figures 1 and 2).
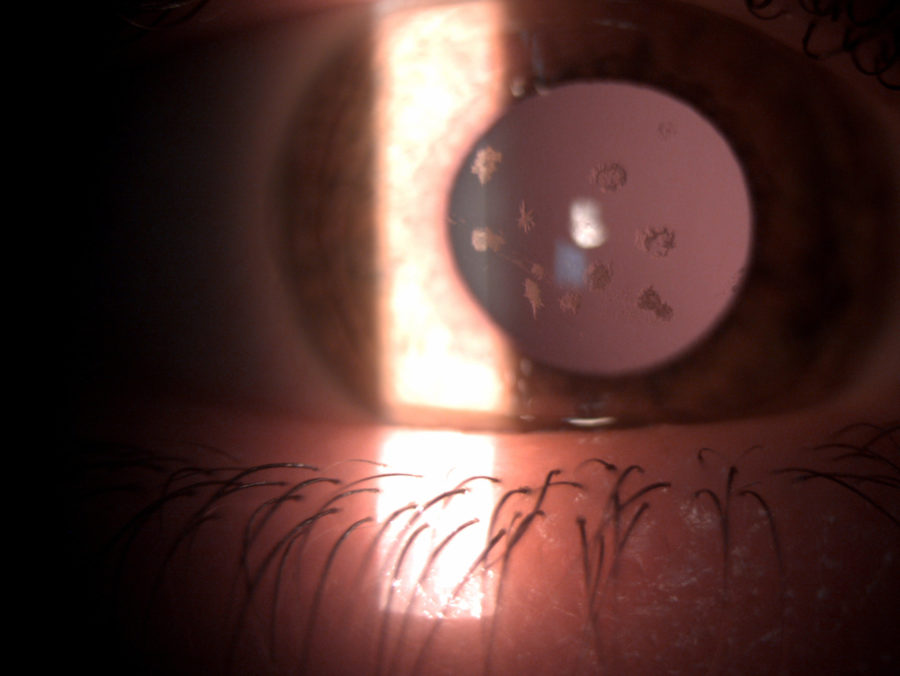
Figure 2: Slit-lamp photograph of the right eye showing whitish, irregularly-shaped, sharply-demarcated granules in the anterior corneal stroma, with clear intervening stroma and sparing corneal periphery.
Anterior segment optical coherence tomography (OCT) demonstrated multiple hyper reflective spots (dots) in the anterior stroma of both corneas, some of them underlining the epithelium on the right eye. Notice that corneal periphery is spared. (Figure 3).
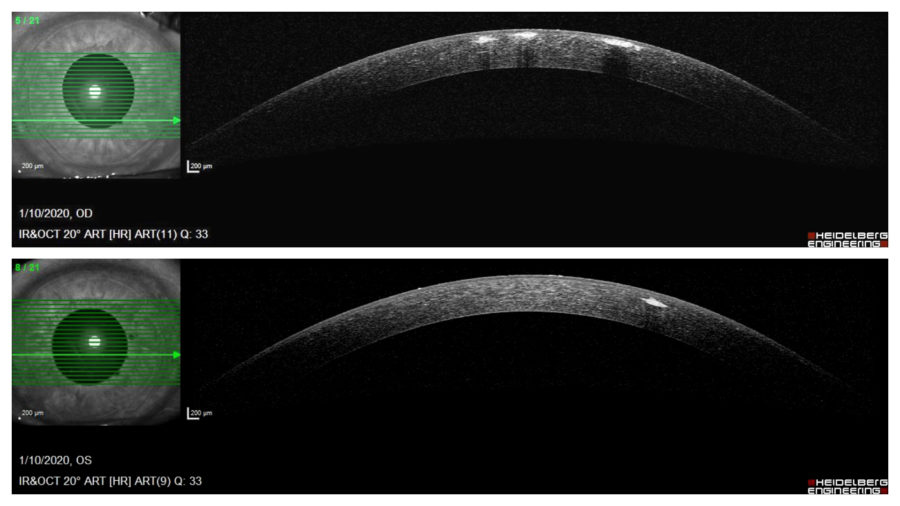
Figure 3: Anterior segment OCT (Heidelberg Engineering®) showing multiple granules in the anterior stroma, more numerous and anteriorly distributed on the right eye. Notice that corneal periphery is spared.
Additional History
The patient had undergone an ophthalmologic examination a couple of years before and was told about the corneal spots on that occasion. Her mother had been examined by the same doctor and was detected with an identical ocular aspect. Neither mother nor daughter suffered from any other ocular or systemic disorder.
Differential Diagnosis
- granular corneal dystrophy (GCD) type I
- other types of corneal dystrophy
- monoclonal gammopathies
- corneal degeneration
- recurrent corneal erosions
- infectious keratitis
- neurotrophic keratitis
- band keratopathy
- diseases of basement membrane (Alport and Usher syndromes)
The major distinction to be done for corneal dystrophies (CD) is with corneal degeneration. Generally, CDs have a bilateral, symmetric presentation, unrelated to environmental factors. Most often, though, CDs must be differentiated among one another, as clinical presentations may overlap. Biomicroscopy helps in identifying different types of CD. Genetic testing may aid in final differentiation. Specifically, lesions in GCD type I can resemble a lot those found in monoclonal gammopathies.
Discussion and Literature
Corneal dystrophy is a collection of rare hereditary, non-inflammatory disorders of abnormal depositions in the cornea. As proposed in 2015 by the International Classification of Corneal Dystrophies (IC3D), CD is subclassified by anatomic location: epithelial/subepithelial, epithelial-stromal, stromal, and endothelial. There is no gender predilection There seems to be a higher prevalence among Caucasian individuals. Clinical manifestation depends on the involved corneal layer(s). Based on the type and severity of CD, patients can be asymptomatic or experience any combination of visual loss, photophobia, dry eye, corneal edema, recurrent corneal erosions and pain. Symptoms can begin at any age and management ranges from simple observation to corneal transplantation.1 The IC3D lists 22 distinct forms of corneal dystrophy, but more than 30 forms have already been described.2 Inheritance is predominantly autosomal dominant, although autosomal recessive and X-chromosomal dominant patterns do exist.3
Formerly called corneal dystrophy Groenouw type I or “classic”, GCD type I belongs to the TGFBI-associated CDs (corneal dystrophies associated with mutations in the transforming growth factor, ß-induced gene).4 Inheritance is autosomal dominant with complete penetrance. It has been well mapped, with MIM (Mendelian Inheritance in Man) #121900, genetic locus 5q31, predominant mutation Arg555Trp.5, 6 In light microscopy, GCD displays multiple stromal deposits, mostly distributed in the anterior stroma. Lesions are seen in transmission electron microscopy as dense rod-shaped bodies and, in confocal microscopy, as hyperreflective opacities in snowflake and trapezoidal shapes. In immunohistochemistry essays, the abnormal material reacts with antibodies to TGFBI protein (keratoepithelin).7
Clinically, GCD type I may be noticeable from childhood, as early as 2 years of age. Glare and photophobia are initial symptoms. At slit-lamp examination, GCD I is characterized by opacities in the corneal stroma, in an irregular crum- or flake-like fashion, slightly white.8 Deposits may be both discrete and confluent, but do not extend to the limbus. In children, a vortex pattern of brownish granules develops, superficial to the Bowman layer. As the patient ages, the size and number of granules increase, resulting in the typical glassy splinters, crushed bread crumbs or snowflakes appearance. Later in life, granules may coarsen and extend into the deeper stroma, approaching Descemet’s membrane.
Management and prognosis of GCD type I depend on stage and manifestations. Homozygote cases have more severe symptoms and the type of mutation is an important phenotype determinant.9 As the disease progresses and the opacities become more confluent in the superficial cornea, reduction in visual acuity (VA) and recurrent erosions occur, requiring medical intervention. Although serial observation demonstrates that granule-drop out after epithelial erosions may actually result in stromal clearing, some patients may need lamellar or penetrating keratoplasty (PKP) by the fifth decade.8 According to deposit size and distribution, an alternative treatment to improve VA and postpone corneal grafting is phototherapeutic keratectomy (PTK).10 However, recurrence-free survival is longer with PKP.11 Refractive corneal surgeries are contraindicated as they induce progression of the disease. Deep anterior lamellar keratoplasty (DALK) is a good therapeutic option, even if repeated procedures are required.12
Keep in mind
- GCD type I is typically an anterior stromal form of CD, but may extend anterior and posteriorly as the disease progresses.
- Despite complete penetrance of the abnormal gene, phenotypic manifestation varies. Interventional therapy is reserved for symptomatic cases.
- Homozygosity is related to higher severity, therefore parental counseling is advisable.
References
- Moshirfar, M., Bennett, P., & Ronquillo, Y. (2021). Corneal Dystrophy. In StatPearls. StatPearls Publishing.
- Munier F., Schorderet D. (2008) Classification of Corneal Dystrophies on a Molecular Genetic Basis. In: Reinhard T., Larkin F. (eds) Cornea and External Eye Disease. Essentials in Ophthalmology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-33681-5_5
- Lisch, W., & Weiss, J. S. (2019). Clinical and genetic update of corneal dystrophies. Experimental eye research, 186, 107715. https://doi.org/10.1016/j.exer.2019.107715
- Solari, H. P., Ventura, M. P., Perez, A. B., Sallum, J. M., Burnier, M. N., Jr, & Belfort, R., Jr (2007). TGFBI gene mutations in Brazilian patients with corneal dystrophy. Eye (London, England), 21(5), 587–590. https://doi.org/10.1038/sj.eye.6702264
- Malkondu, F., Arıkoğlu, H., Erkoç Kaya, D., Bozkurt, B., & Özkan, F. (2020). Investigation of TGFBI (transforming growth factor beta-induced) Gene Mutations in Families with Granular Corneal Dystrophy Type 1 in the Konya Region. Turkish journal of ophthalmology, 50(2), 64–70. https://doi.org/10.4274/tjo.galenos.2019.55770
- Eiberg, H., Møller, H.U., Berendt, I. et al. Assignment of Granular Corneal Dystrophy Groenouw Type I (CDGG1) to Chromosome 5q. Eur J Hum Genet 2, 132–138 (1994). https://doi.org/10.1159/000472353
- Weiss, J. S., Møller, H. U., Aldave, A. J., Seitz, B., Bredrup, C., Kivelä, T., Munier, F. L., Rapuano, C. J., Nischal, K. K., Kim, E. K., Sutphin, J., Busin, M., Labbé, A., Kenyon, K. R., Kinoshita, S., & Lisch, W. (2015). IC3D classification of corneal dystrophies–edition 2. Cornea, 34(2), 117–159. https://doi.org/10.1097/ICO.0000000000000307
- Lin, Z. N., Chen, J., & Cui, H. P. (2016). Characteristics of corneal dystrophies: a review from clinical, histological and genetic perspectives. International journal of ophthalmology, 9(6), 904–913. https://doi.org/10.18240/ijo.2016.06.20
- Møller H. U. (1989). Inter-familial variability and intra-familial similarities of granular corneal dystrophy Groenouw type I with respect to biomicroscopical appearance and symptomatology. Acta ophthalmologica, 67(6), 669–677. https://doi.org/10.1111/j.1755-3768.1989.tb04400.x
- Seitz, B., Behrens, A., Fischer, M., Langenbucher, A., & Naumann, G. O. (2004). Morphometric analysis of deposits in granular and lattice corneal dystrophy: histopathologic implications for phototherapeutic keratectomy. Cornea, 23(4), 380–385. https://doi.org/10.1097/00003226-200405000-00013
- Lewis, D. R., Price, M. O., Feng, M. T., & Price, F. W., Jr (2017). Recurrence of Granular Corneal Dystrophy Type 1 After Phototherapeutic Keratectomy, Lamellar Keratoplasty, and Penetrating Keratoplasty in a Single Population. Cornea, 36(10), 1227–1232. https://doi.org/10.1097/ICO.0000000000001303
- Uit de Bosch, L. M., Ormonde, S., & Misra, S. L. (2016). Visual Outcomes Following Deep Anterior Lamellar Keratoplasty in Granular Corneal Dystrophy Types 1 and 2. Korean journal of ophthalmology : KJO, 30(6), 481–482. https://doi.org/10.3341/kjo.2016.30.6.481
