Presented by: Miltos Balidis MD, PhD, FEBOphth, ICOphth

Edited by: Penelope Burle de Politis, MD

A 25-year-old woman was referred for investigation of long-standing bilateral ocular surface inflammation.
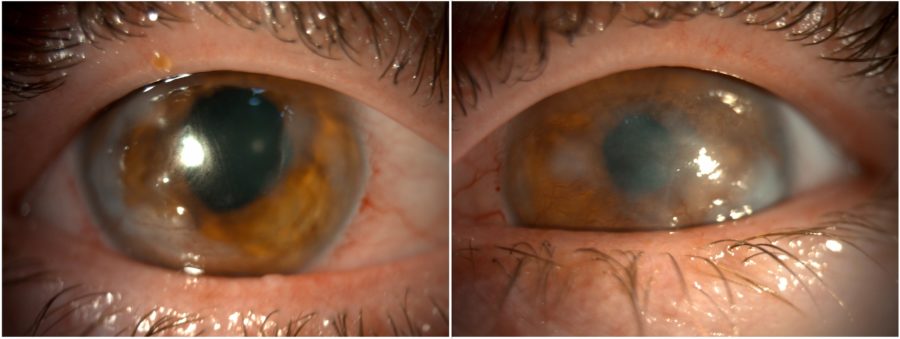
Figure 1: Slit-lamp photograph showing central and peripheral corneal opacities, corneal vascularization, and an irregular pupil in both eyes.
Case History
A 25-year-old Caucasian woman was referred for diagnosis and management of chronic ocular surface inflammation in both eyes (BE). She had been under different topical corticosteroid-antibiotic combinations over the years, without any significant or long-lasting relief. Upon ophthalmological examination, her corrected distance visual acuity (CDVA) was 1/10 in the right eye (RE) and 0.4/10 in the left eye (LE). Refractometry was +0.25 -1.75@90° in the RE and +1.00 -1.50@10° in the LE, and intraocular pressure was respectively 11 and 12 mmHg. A bilateral, low-amplitude, high-frequency nystagmus was observed. Under biomicroscopy, BE featured central and peripheral elevated, grayish-white corneal opacities, corneal vascularization, and a slightly irregular pupil (Figure 1). Meibomian gland disorder (MGD), papillae at the tarsal conjunctiva, and a grade 1 peripheral nuclear cataract were detected in BE (Figure 2). Fundoscopy was impracticable due to the nystagmus and corneal opacities.
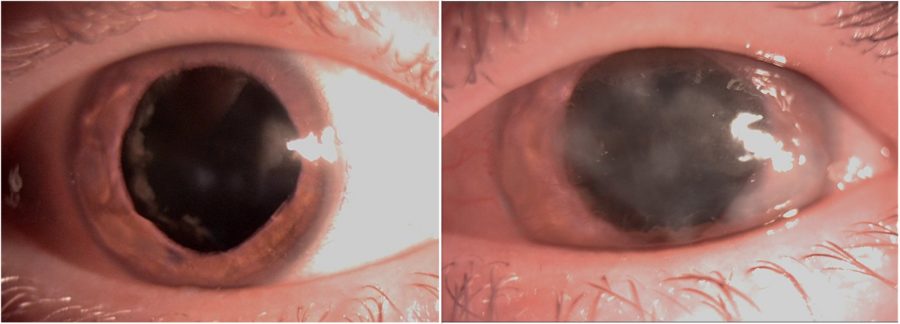
Figure 2: Slit-lamp photograph of both eyes under mydriasis illustrating the peripheral nuclear cataract and the extension of the corneal opacities in both eyes.
The initial management strategy targeted ocular surface inflammation and consisted of omega-3 fatty acids and doxycycline p.o., intense regulated pulsed light (IRPL), topical and subconjunctival corticosteroids, and non-preserved artificial tears, resulting in both subjective and objective improvement. Once the eyes were calm, bilateral excimer laser phototherapeutic keratectomy (PTK) was performed to remove scarring tissue and enhance corneal regularity. The procedure led to an improvement in visual acuity to 2/10 (RE) and 1/10 (LE) and an important reduction in photophobia. Additionally, there was an increase in corneal transparency and considerable shrinkage of the corneal vessels in the RE (Figure 3).
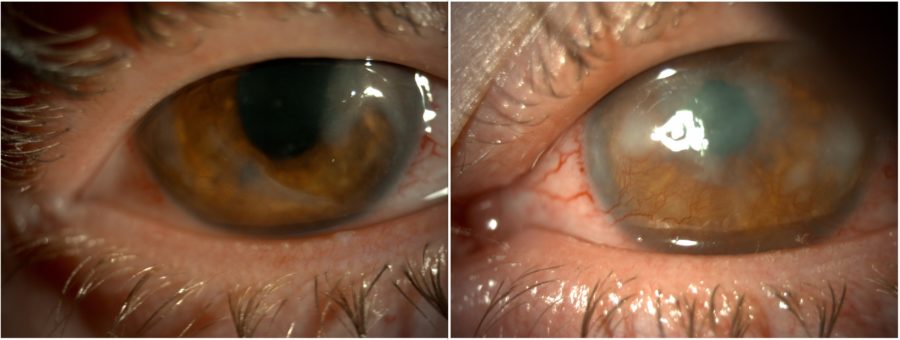
Figure 3: Post-PTK slit-lamp photograph of both eyes displaying an improvement in corneal transparency and less prominent corneal vessels in the RE.
Additional History
When inquired about her family history, the patient informed that her sister and mother suffered from the same ocular conditions, namely nystagmus and low visual acuity since birth. They were called in for examination and presented identical ocular characteristics, particularly subepithelial scarring and sectoral iris atrophy (Figures 4 and 5).
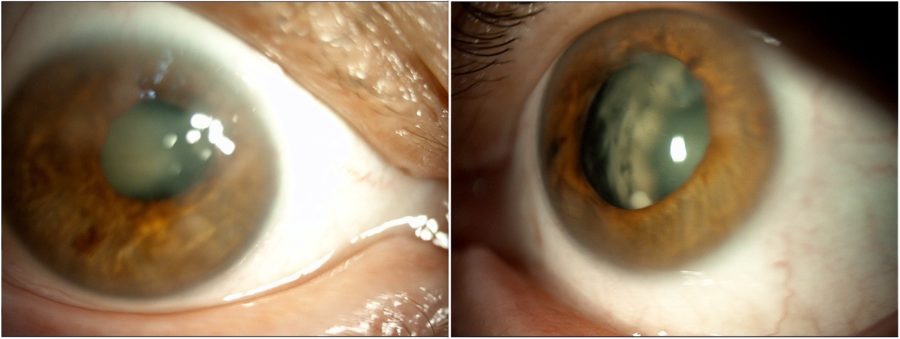
Figure 4: Slit-lamp photograph of the patient’s sister exhibiting a remarkably analogous pattern of corneal opacities and lens sclerosis in BE.
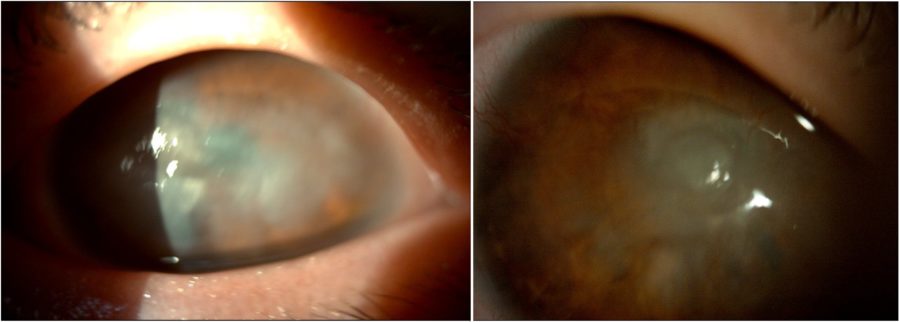
Figure 5: Slit-lamp photograph of the patient’s mother depicting anterior segment changes that closely mirror those seen in both her daughters’ eyes.
Among all three relatives, fundus imaging was only feasible for the patient’s RE, which had fewer central opacities compared to the other 5 eyes involved in the investigation. Spectral domain optical coherence tomography (SD-OCT) revealed a totally undeveloped foveal pit (Figure 6).
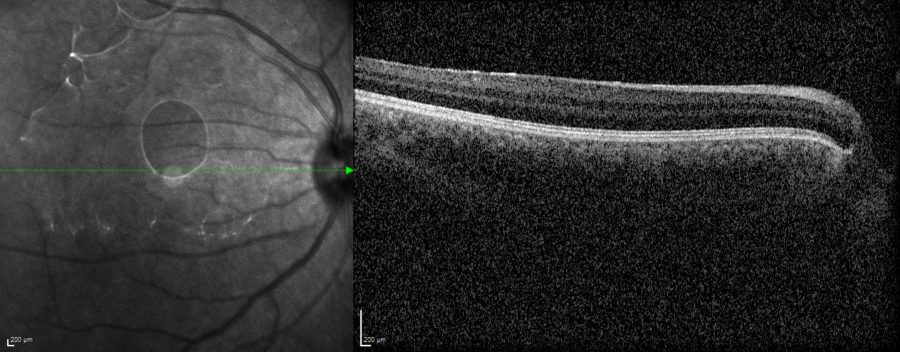
Figure 6: Spectral domain optical coherence tomography (SD-OCT, Heidelberg Engineering®) of the patient’s RE demonstrating complete foveal aplasia.
An electrophysiology study and genetic testing were requested for the three family members. The full-field electroretinogram (ffERG) displayed a similar pattern in all of them, with subnormal to abnormal responses in BE (Figures 7 and 8).
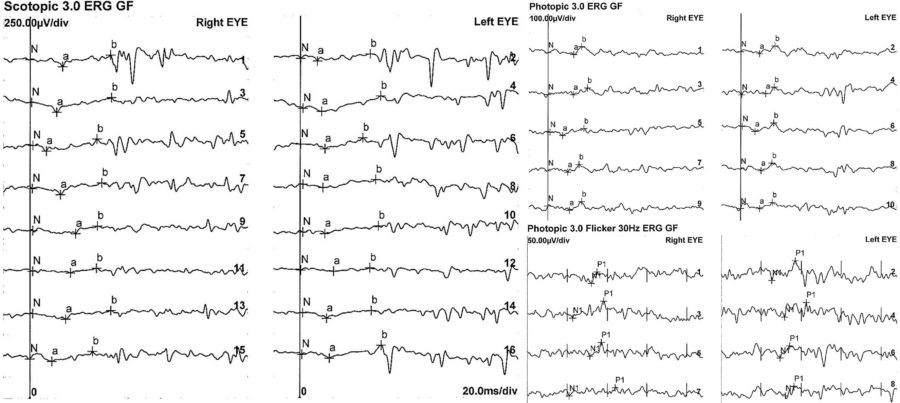
Figure 7: Patient’s full-field ERG. Scotopic ffERG: rod-specific b-wave within normal limits. Bright-flash dark-adapted ERG a- and b-wave peak times bilaterally normal in BE. Photopic ffERG: 30Hz flicker ERG and single-flash ERG mildly reduced and delayed bilaterally.
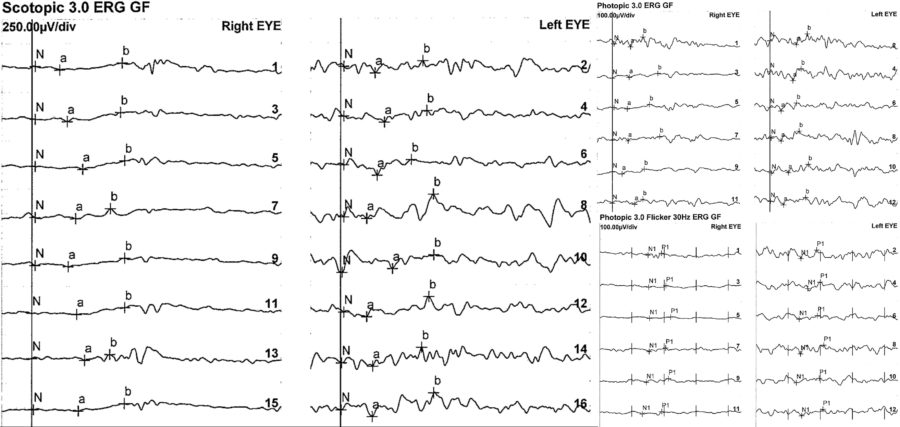
Figure 8: Patient’s sister’s full-field ERG exhibiting even more expressively depressed responses, especially in the RE.
Genetic testing confirmed the diagnostic hypothesis of PAX6-related ocular dysgenesis.
Satisfactory long-term control of surface inflammation was achieved with topical administration of cyclosporine and low-dose topical corticosteroid drops, and frequent ocular lubrication. Visual acuity remains stable in BE. Genetic counseling was conducted with the women in reproductive age.
Differential Diagnosis of PAX6-related Ocular Dysgenesis
- Salzmann’s Nodular Degeneration (SND)
- Schnyder crystalline corneal dystrophy (SCCD)
- Limbal Stem Cell Deficiency (LSCD)
- Peters’ anomaly
- Rieger’s anomaly
- ocular albinism
- retinal dystrophy
The large spectrum of phenotypes in PAX6-related ocular dysgenesis make its diagnosis a challenge. The presence of nystagmus with or without aniridia, and the occurrence in other family members must raise suspicion of the disorder as genetic testing and counseling are crucial steps in management.
Discussion and Literature
The transcription factor PAX6 is essential in ocular development in vertebrates, being considered the master regulator of ocular and neural development. Years after its identification, PAX6 is one of the most intensively studied genes, both in terms of its diverse and complex functions during oculogenesis and its role in an ever-increasing variety of congenital ocular defects. PAX6 mutations are archived in the Human PAX6 Allelic Variant Database, which currently contains hundreds of records, most of which are related to the eye.
The most common of these malformations is aniridia, but PAX6 mutations also cause a range of non-aniridia phenotypes such as optic nerve defects, keratitis, microphthalmia, and foveal hypoplasia. Non-aniridia phenotypes are frequently associated with missense mutations. PAX6-related mutations result in under- or abnormal development of eye structures including the cornea, leading to a bilateral and progressive limbal stem cell insufficiency and conjunctivalization of the cornea.
Another manifestation of PAX6-related ocular dysgenesis is foveal hypoplasia (FVH), characterized by an underdeveloped foveal depression with a relatively preserved neuroretina. FVH can be isolated (IFVH), infantile nystagmus being the presenting sign. However, it is most commonly accompanied by other ocular or systemic diseases, such as ocular or oculocutaneous albinism, aniridia, optic nerve hypoplasia, achromatopsia, or Stickler syndrome. A full iris with subtle structural abnormalities is more common in patients with PAX6-associated IFVH.
There are two forms of IFVH: foveal hypoplasia 1, with or without anterior segment anomalies and/or cataract (FVH1) – autosomal dominant mode of inheritance –, and foveal hypoplasia 2, with or without optic nerve misrouting and/or anterior segment dysgenesis (FVH2) – an autosomal recessive disorder. The two phenotypes of FVH1 are: isolated foveal hypoplasia, and foveal hypoplasia with presenile cataract and peripheral corneal pannus. The phenotypes of FVH2 include optic nerve misrouting, and the presence of posterior embryotoxon, Axenfeld anomaly, and coloboma. Both FVH1 and FVH2 have been associated with low visual acuity and nystagmus.
Infantile nystagmus is a genetically heterogeneous disorder consisting of involuntary oscillations of the eyes, often manifested within the first 6 months of life. Congenital nystagmus (CN) shows variable clinical features. PAX6 mutations can also cause autosomal-dominant nystagmus, typically in association with aniridia or iris hypoplasia, but also without aniridia, thus the importance of screening for the PAX6 gene in patients who present with CN.
The clinical phenotypes obtained as a result of the PAX6 gene mutations are complicated and vary among patients, even among those who carried the same variants. Molecular diagnosis may be helpful in determining the etiology of the corresponding disorders with complicated and occult features. The phenotypic spectrum caused by PAX6 variants suggests an overestimation of sporadic cases with PAX6 variants, with strong implications for both genetic counseling and family planning.
Keep in mind
- Corneal opacities accompanied by nystagmus should raise suspicion for associated posterior pole abnormalities, possibly congenital or genetic.
- PAX6-related ocular dysgenesis is commonly correlated with aniridia, but not always.
- Because of the inheritable trait of PAX6-related ocular dysgenesis, genetic testing is crucial in suspected cases and reproductive counseling is advised.
References
- Lima Cunha D, Arno G, Corton M & Moosajee M (2019). The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes, 10(12), 1050. https://doi.org/10.3390/genes10121050
- Hanson IM (2003). PAX6 and congenital eye malformations. Pediatric research, 54(6), 791–796. https://doi.org/10.1203/01.PDR.0000096455.00657.98
- Tzoulaki I, White IM & Hanson IM (2005). PAX6 mutations: genotype-phenotype correlations. BMC genetics, 6, 27. https://doi.org/10.1186/1471-2156-6-27
- Lizhu Yang, Qi Zhou, Zixi Sun, Kaoru Fujinami, Huajin Li, Zhisheng Yuan, Kazuo Tsubota & Ruifang Sui (2017). Phenotype and Genotype Features of PAX6-related Ocular Dysgenesis. Invest. Ophthalmol. Vis. Sci. 2017;58(8):577. https://iovs.arvojournals.org/article.aspx?articleid=2638735
- Lagali N, Wowra B, Fries FN, Latta L, Moslemani K, Utheim TP, Wylegala E, Seitz B & Käsmann-Kellner B (2020). PAX6 Mutational Status Determines Aniridia-Associated Keratopathy Phenotype. Ophthalmology, 127(2), 273–275. https://doi.org/10.1016/j.ophtha.2019.09.034
- Jiang Y, Li S, Xiao X, Sun W & Zhang Q (2021). Genotype-Phenotype of Isolated Foveal Hypoplasia in a Large Cohort: Minor Iris Changes as an Indicator of PAX6 Involvement. Investigative ophthalmology & visual science, 62(10), 23. https://doi.org/10.1167/iovs.62.10.23
- Matsushita I, Morita H & Kondo H (2020). Autosomal dominant foveal hypoplasia without visible macular abnormalities and PAX6 mutations. Japanese journal of ophthalmology, 64(6), 635–641. https://doi.org/10.1007/s10384-020-00766-9
- Lee B, Choi DG, Chun BY, Oh EH, Lee YJ, Kim UK & Park JS (2019). A family with a mild form of congenital nystagmus and optic disc coloboma caused by a novel PAX6 mutation. Gene, 705, 177–180. https://doi.org/10.1016/j.gene.2019.04.035
- Thomas S, Thomas MG, Andrews C, Chan WM, Proudlock FA, McLean RJ, Pradeep A, Engle EC & Gottlob I (2014). Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. European journal of human genetics : EJHG, 22(3), 344–349. https://doi.org/10.1038/ejhg.2013.162
- Huang L, Peng J, Xie Y, Zhou Y, Wang X, Wang H, Gui J & Li N (2023). Diversity of clinical phenotypes in a cohort of Han Chinese patients with PAX6 variants. Frontiers in genetics, 14, 1011060. https://doi.org/10.3389/fgene.2023.1011060
- Lima Cunha D, Owen N, Tailor V, Corton M, Theodorou M & Moosajee M (2021). PAX6 missense variants in two families with isolated foveal hypoplasia and nystagmus: evidence of paternal postzygotic mosaicism. European journal of human genetics : EJHG, 29(2), 349–355. https://doi.org/10.1038/s41431-020-00737-1
